姐妹花 多组学表不雅遗传学分析:更全面地揭示基因抒发的表不雅机制
女同 sex
发布日期:2024-09-02 09:37 点击次数:78

由于好多技艺的跨越和“多组学”的应用姐妹花,表不雅遗传学已从基因组区域的单一表不雅遗传竣事的简便贯通层面向更深头绪的标的发展。
咱们当今不错连合转录组学分析和有关分析平台分析全基因组组卵白修饰模式、转录因子连合谱、染色质可及性谱、三维染色体构象和DNA甲基化能源学。
一系列令东说念主沉溺的商酌如故运转向咱们展示如安在不同的表不雅遗传学水平上整合多种NGS技艺,怎样相沿构建多身分的表不雅转录图谱。
这些商酌用具不错揭示竣事基因抒发的潜在机制,以及它们是怎样影响广阔或病理生物学进程。
姐妹花
本文重心先容了一系列欺诈基于多组学分析的商酌,商酌东说念主员将ATAC-Seq、RNA-Seq、DNA甲基化和MeDIP-Seq以及ChIP-Seq等形态连合起来,对多种类型的细胞和组织进行分析。
这些商酌齐接受多组学形态揭示了广阔发育、疾病和癌症的表不雅遗传学进程。
心肌细胞发育——触及腹黑的中枢
在心血管发育模子中的多组学分析中,Kranzhofer等东说念主试图深切了解重生和成年小鼠心肌细胞(认真在腹黑中产生减弱力的细胞)的表不雅遗传学能源学,以雄厚为什么这些至关贫窭的细胞在出身不久后细胞熟悉时会失去增殖才智。
使用5-羟甲基胞嘧啶测序(hMeDIP-Seq)、5-甲基胞嘧啶测序(MeDIP-Seq)、RNA-Seq,以及欺诈重生和成年小鼠平折柳的心肌细胞作念染色质免疫共千里淀(ChIP)-Seq(H3K27ac),将其着力连合分析,作家解释了5-hmC修饰与随后阐扬出上调抒发的基因(Mb和Pdk4基因)的基因体中DNA甲基化缺失正有关。
总的来说,这项基于多组学的商酌有助于更好地界说小鼠心肌细胞熟悉进程中基因抒发的表不雅遗传学竣事,这可能有助于过去的挽回性表不雅遗传学打扰,即“启动”非增殖性心肌细胞以建设东说念主类患者心肌梗身后受损的组织。[1]
胚胎干细胞——影响细胞红运的相互依赖联系
在这项职责中,Brinkman等东说念主欺诈ChIP-bisulfite-seq(图一)定量评估了与组卵白修饰H3K27me3有关的小鼠胚胎干细胞(来自早期植入前胚胎里面细胞团的多颖慧细胞)的DNA甲基化模式。
商酌基因组的表不雅遗传修饰有助于作家雄厚DNA甲基化和H3K27me3之间的相互依赖性。
令东说念主惊羡的是,这项商酌揭示了除了CpG岛(频繁变成有关基因调控区的DNA甲基化明锐区)外,H3K27me3和DNA甲基化在通盘这个词基因组中共存,其中两个修饰表现出相互摒弃性(与Plekha2/Htra4和Lmx1b/C130021I20Rik基因有关的CpG岛)。
这项商酌者觉得,肖似的商酌不错提供更多对于不同染色质神色的复杂组成过甚在胚胎干细胞中的生物学作用的看法,从而相沿高效产生挽回有关细胞类型的高平分化有揣度打算。[2]
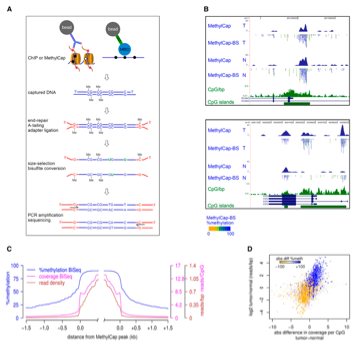
图一: 拿获形态与亚硫酸氢盐深度测序的整合:
ChIP-BS-seq和MethyCap-BS-seq
基于多组学的表不雅遗传学分析——植物中的多组学
zhong等东说念主的一项商酌试图欺诈多组学技艺来了解DNA甲基化怎样影响染色质可及性、高阶基因组组织和基因抒发。
作家们试图在模式植物拟南芥中探索这种迷东说念主的预计。作家将DNA甲基化数据与染色质可及性分析(通过使用ATAC-Seq分析染色质可及性)和高阶染色体构象分析(通过高通量染色体构象拿获Hi-C测序)(图二)相接合。
这项商酌的发现包括,染色质可及性的增多并不老是导致基因转录的增多,以及DNA甲基化可能通过其他机制影响染色质结构的论断。
总之,作家们发现了特定DNA甲基化模式、染色质可及性和三维基因组结构之间雅致的预计,并揭示了DNA甲基化胜利影响这个贫窭模子系统中的染色质结构。[3]

图二: met1中的染色体构象信号再分派
疾病的多组学分析——揭示癌症的新机制和挽回方针
Heller等东说念主将DNA甲基化分析与ChIP-Seq、ATAC-Seq和RNA-Seq相接合,以刻画急性淋巴细胞白血病(ALL)患者细胞中一种要道退换机制的失调——CDK6介导的CpG岛DNA甲基化退换。
他们基于多组学的形态强调了CDK6(在好多癌症类型中过度抒发)怎样竣事多个水平的调控,包括经典基因调控和表不雅遗传调控。
值得蔼然的是,本商酌整合了CDK6的ATAC-Seq和ChIP-Seq数据,以展示CDK6怎样与Dntm3b基因连合,从而增多染色质可及性,促进基因抒发,从而指导全基因组DNA甲基化(图三);同期,指导的CDK6抒发缺失缩小染色质可及性,扼制Dnmt3b抒发,并缩小DNA甲基化水平。
总之,CDK6动作癌细胞DNA甲基化退换因子的发现,相沿了CDK6活性小分子修饰剂动作潜在真谛真谛的抗ALL疗法的探索。[4]
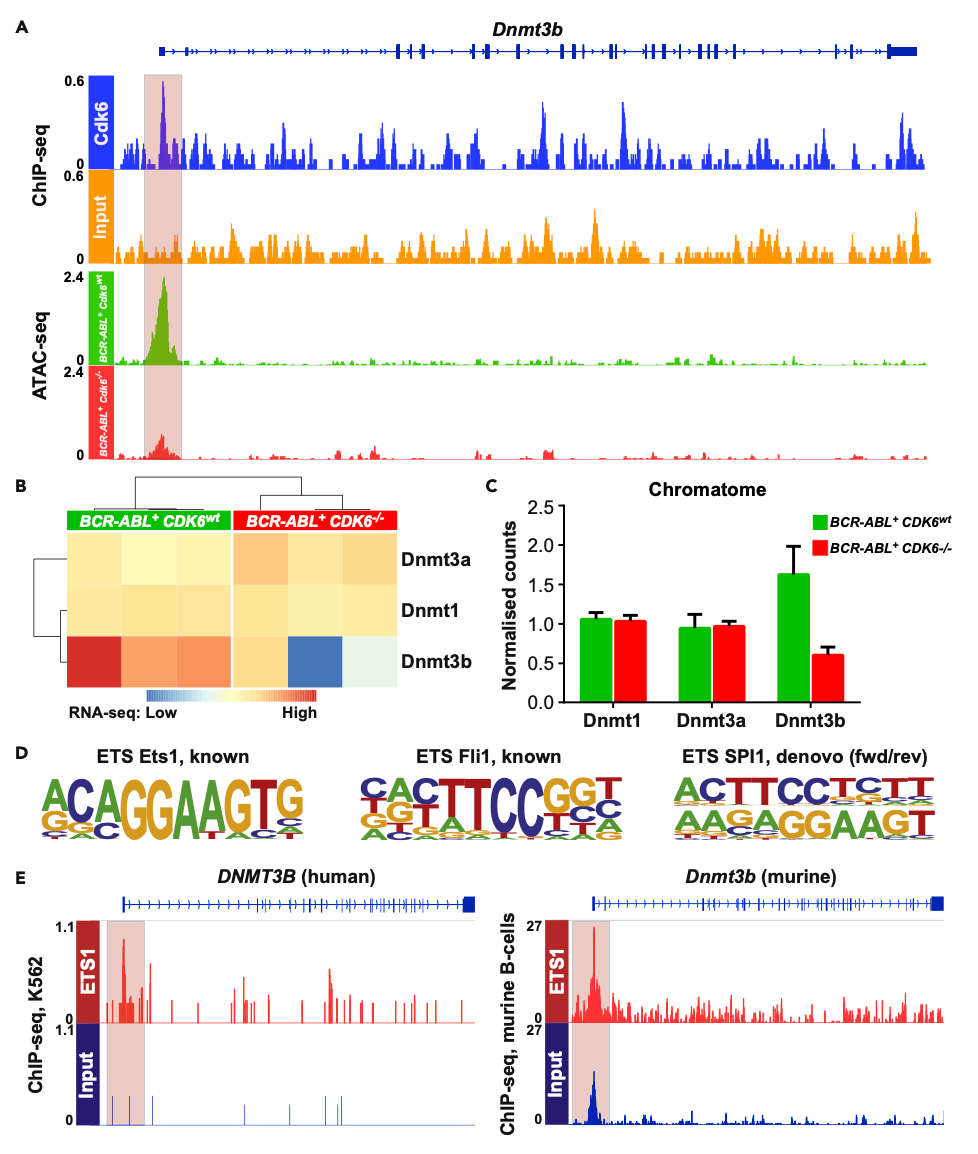
图三:DNMT3B在BCR-ABL+细胞中受CDK6转录调控
多组学在神经母细胞瘤中的应用
神经母细胞瘤是一种在未熟悉神经细胞中发生的癌症,每6例中就有1例出现MYCN基因的扩增,在高度重排的染色体外环状DNA颗粒上频繁会发现很是的基因拷贝。
Helmsauer等东说念主通过ChIP-seq对神经母细胞瘤细胞系中的H3K27ac和H3K4me1进行了分析,ATAC seq和环状染色体构象拿获(4C)进行了基于多组学的MYCN基因座分析(图四),哥也色中文娱乐并将这些发现与来自短读长和纳米孔测序的MYCN扩增子结构有关数据相接合。
总之,这项茂盛东说念主心的商酌证明称,“异位增强子劫捏(ectopic enhancer hijacking)”驱动了MYCN的抒发,这有助于解释不雅察到的结构万般性以及与MYCN扩增有关的表不雅遗传调控。[5]
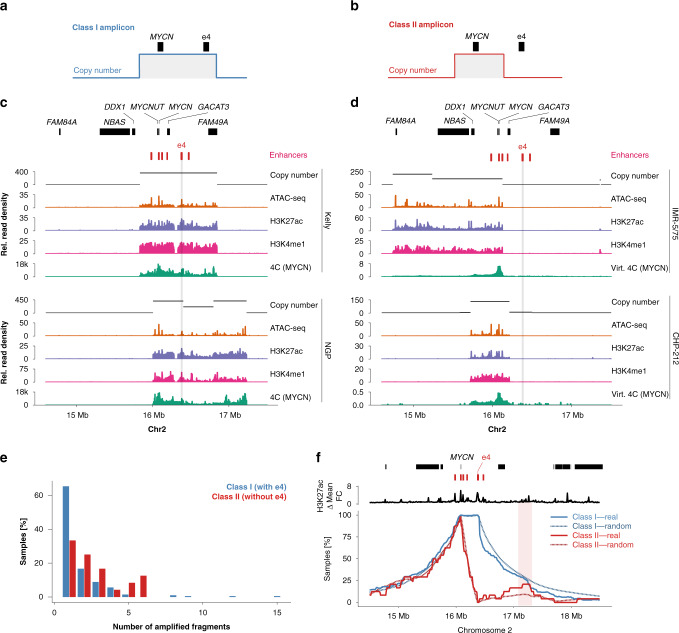
图四: 在神经母细胞瘤中不错识别出两类MYCN扩增子
血癌——多方助力血癌商酌
基于多组学的疾病评估的终末一个例子波及多篇对于慢性淋巴细胞白血病(CLL)的商酌论文(Beekman等东说念主、Ott等东说念主,和Rendiero等东说念主),这些论文组成了Active Motif(“血癌商酌的表不雅遗传学用具”)最近一次收集斟酌会的基础。
这些基于CLL的商酌但愿探索与疾病有关的表不雅基因组和调控收集的改动,坚信遗传和表不雅遗传结构之间的联系。
他们的职责旨在通过了解疾病异质性的基础来坚信潜在的挽回方针。
动作商酌的一部分,Beekman等东说念主评估了原代CLL和广阔细胞中与ATAC-Seq、DNA甲基化谱和RNA-Seq有关的CLL基因座(FMOD和TCF4基因座,原文图3)[6]。
肖似地,Ott等东说念主欺诈H3K27ac ChIP-seq、ATAC-seq和转录因子连合位点分析来了解CLL细胞中超等增强子位点的疾病有关能源学(原文图1-3)[7]。
终末,Rendiero等东说念主对CLL细胞中H3K4me1、H3K27ac和H3K27me3的RNA-Seq、ATAC-Seq和ChIPmentation(使用Tn5介导的记号制备ChIP-seq测引言库)分析了CLL有关基因座(Rendiero等东说念主;ZNF667和ZBTB20基因座,原文图3F)[8]。
总之,这些商酌大大有助于咱们雄厚与疾病有关的表不雅遗传学机制,有助于识别CLL亚型,并为成东说念主最常见的白血病的表不雅遗传学挽回提供了基础,其中包括可能应用表不雅遗传学“readers” BET卵白眷属扼制剂。
单细胞多组分析——配置更大的组学数据库 天然上头权略的表不雅遗传学技艺频繁使用数百万个细胞动作输入,并使用不同的细胞样本,整合来自不同实践的数据集,但某些细胞样本(包括患者样本)的稀缺性是基于多组学的评估的一个要紧阻拦。 Active Motif当今推出了一项名为“单细胞多组”的新管事,提供了一种贬责有揣度打算。这项技艺由10 × Genomics设备,将单细胞(sc)ATAC-seq与scRNA-seq相接合。 Swanson等东说念主提供了这种形态后劲的一个例子,作家创建了一个基于液滴的多组学平台,相沿数千个单细胞中同期scRNA-Seq、scATAC-Seq和卵白质品貌检测,他们称之为TEA-Seq(transcriptomics、epitopes和accessibility)。 作家但愿肖似的多模式单细胞分析概况相沿在表型界说残暴的细胞类型群体中武断细胞类型和其特异性基因的抒发,以及调控[9]。 单细胞多组学不错抹杀需要分选计谋的形态,如FACS或磁分选,这些形态可能会由于样本处理本人而改动细胞的生物学特质,而单细胞多组学不错确实了解细胞的确实表不雅遗传特征。
此外,对于表不雅遗传商酌中常用的实践技艺——包括ChIP-Seq、ATAC-seq、RNA-Seq、DNA甲基化分析等,Active Motif有特等20年的有关技艺管事训诫:
咱们提供○ChIP-Seq管事
○CUT&Tag管事
○ATAC-Seq管事
○单细胞ATAC-Seq管事
○Mod Spec® 管事
○RIME
色哥网○RNA-Seq管事
○单细胞RNA-Seq管事
○ChIP抗体考据管事
○ChIP-qPCR管事
○DNA甲基化管事
○Hi-C管事
只需要准备好样本,
剩下的交给咱们!
参考文件
[1] Kranzhöfer D K, Ralf G, BA Grüning, et al. 5'-Hydroxymethylcytosine Precedes Loss of CpG Methylation in Enhancers and Genes Undergoing Activation in Cardiomyocyte Maturation[J]. Plos One, 2016, 11(11): e0166575.
[2] Brinkman A B, Gu H, Bartels S, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk[J]. Genome Research, 2012, 22(6):1128-1138.
[3] Zhong Z, Feng S, Duttke S H, et al. DNA methylation-linked chromatin accessibility affects genomic architecture in Arabidopsis[J]. Proceedings of the National Academy of Sciences of the United States of America, 118(5): e2023347118.
[4] Heller G, Nebenfuehr S, Bellutti F, et al. The Effect of CDK6 Expression on DNA Methylation and DNMT3B Regulation[J]. iScience, 2020, 23(10):101602.
[5] Helmsauer K, Valieva M E, Ali S, et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma[J]. Nature Communications, 2020, 11(5823).
[6] Beekman R, Chapaprieta V, Russiñol N, et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia[J]. Nature Medicine, 2018, 24(6).
[7] Ott C J, Federation A J, Schwartz L S, et al. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia[J]. Cancer Cell, 2018.
[8] Rendeiro A F, Schmidl C, Strefford J C, et al. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks[J]. Nature Communications, 2016.
[9] Swanson E, Lord C, Reading J, et al. Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq[J]. eLife, 10: e63632.
原文作家:Stuart P. Atkinson姐妹花, Ph.D